Step 3: ENTER SAMPLE INFORMATION
Step 4: CONFIGURE YOUR ANALYSIS RUN
Step 5: RUNNING AND COMPLETION
1.2.2 Uploading a custom library
2 Description of the PinAPL-Py Analysis output
1 Running PinAPL-Py
1.1 QUICK START:
Step 1: SET UP A RUN
Enter a project name for your analysis run. This name will help you identify your results in case you do multiple runs in a row. Provision of an email address is optional, but will let you safely close the browser during the analysis and receive a notification after completion.
Step 2: UPLOAD DATA
Upload your files via the drag-and-drop frame. Uncompressed format (.fastq) is supported, but compressed (.fastq.gz) is strongly recommended for speed.
Step 3: ENTER SAMPLE INFORMATION
Enter the name of the condition each file represents. Files representing replicates of the same condition have to be given the same name. Do not number your replicates. Numbering is done automatically by the program and displayed on the results page after completion of the analysis.
Please mark all control replicates with the checkbox to the right. Naming the control samples is optional
Example: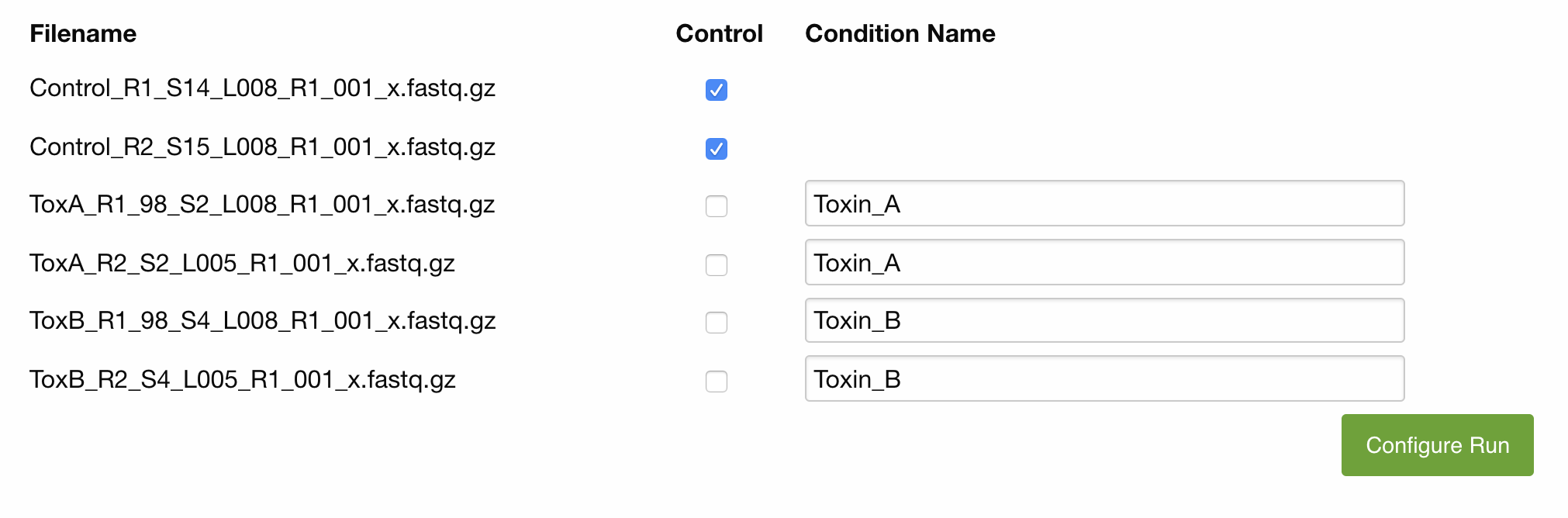
Step 4: CONFIGURE YOUR ANALYSIS RUN
First, choose the screen type. Choose between “enrichment” (e.g. a drug resistance screen) or “depletion” (e.g. a gene-essentiality screen), depending on whether your screen aims at finding sgRNAs of high or low abundance, respectively. Next, choose the sgRNA library used in your screen from the dropdown menu. If your screen uses a library not present in the list or a custom library, see “Uploading a custom library” in the Advanced Options below.
Optional: If you would like to edit the default parameter settings, click Advanced Options. For instructions on these parameters, see “Parameter description” in the Advanced Options section.
Step 5: RUNNING AND COMPLETION
You can follow the program’s execution log by refreshing the page repeatedly. In case another run was started before yours, your run will be queued and start after completion of the previous.
If you provided an email address, you can close the browser; you will be notified by email and sent a link to the results after completion. Otherwise, please leave the progress screen open.
The results will remain on the server for 5 days. You can download all content shown on the results page in a single ZIP archive.
1.2 ADVANCED OPTIONS
1.2.1 Parameter Description
Gene Ranking
Gene Ranking Metric (default = "SUMLFC"):
Method to combine the sgRNA enrichment/depletion data for ranking of genes:
- SUMLFC: *** NEW in v2.9 *** This method computes a gene score by taking the sum of the log fold-changes of all sgRNAs targeting it, and multiplying it by the number of its sgRNAs that reached statistically significant enrichment/depletion.
- AVGLFC: This method computes a gene score by averaging the log fold-changes of all sgRNAs targeting it.
- αRRA: Adjusted robust rank aggregation (Li et al., 2014). This method computes a gene score based on a Beta model of the aggregation of sgRNAs.
- STARS: STARS score (Doench et al., 2016). This method computes a gene score based on a binomial model. It requires a gene to have at least two sgRNAs ranked among the top x% (see the “sgRNAs Included (STARS only)” parameter below).
For more details on these methods, please refer to the original publications or use our contact page.
Number of permutations (default = 1000):
Number of permutations for p-value estimation of the gene ranking score. CAUTION: If STARS is chosen, reducing the number of permutations (to e.g. 10) is strongly recommended, due to the increased computational demand.
Significance level (default = 0.01)
Significance threshold for gene ranking.
%sgRNAs Included (STARS only) (default = 10):
Percentage of sgRNAs to be included in the ranking analysis. Only relevant if “STARS ”method is chosen.
sgRNA Ranking
p-value adjustment (default = "Sidak"):
Method for p-value adjustment for multiple tests.
- sidak: Sidak correction method.
- fdr_bh: Benjamini-Hochberg correction method.
- bonferroni: Bonferroni correction method.
Significance level (default = 0.01)
Significance threshold for sgRNA ranking.
Cluster by… (default = "Highest Variance"):
Criterion for sample clustering.
- Highest Variance: Clustering of the samples is based on the sgRNAs with the highest read count variance across all samples.
- Highest Counts: Clustering of the samples is based on the sgRNAs with the highest/lowest abundance (depending on whether the screen type is "enrichment"/"depletion").
Top% of sgRNAs for clustering (default = 25):
Specifies what quantile of sgRNAs clustering is based on. In case of clustering by highest counts, the top x sgRNAs from each sample are combined.
Read Counting
Normalization: (default = ‘cpm ’):
Method of read count normalization.
- total: Read counts for each sgRNA are divided by the number of total number of reads in the sample and multiplied by the mean total number of read counts across all samples.
- cpm: Counts per million. Read counts for each sgRNA are divided by the number of total read counts in the sample and multiplied by 1,000,000.
- size: Read counts are normalized using median ratios and the "size-factor" method, as decribed in (Li et al., 2014; Anders and Huber, 2010).
Cutoff (default = 0):
Cutoff threshold (given in cpm) to filter out low sgRNA read counts. sgRNAs with read counts lower than the cutoff will be set to 0.
Round Counts (default = No):
Round read counts after normalization to avoid fractional read counts. Rounding only affects visualization, but not significance analysis.
Averaging (default = Median):
Method of averaging sgRNA read counts across replicates (Median / Mean)
Alignment
sgRNA Length (default = 20)
Length of the sgRNA in the sequence read.
Adapter Error Tolerance (default = 0.1)
Error rate (mismatches and indels) allowed for the identification of the 5’ adapter (Refer to the cutadapt manual for more details). Increasing this rate can help to control for poor sequence quality.
Matching Threshold (default = 40)
Minimal alignment score required to consider a read successfully matched. For a perfect match this must be double the sgRNA sequence length (Refer to the Bowtie2 manual for more details on calculation of the alignment score). Decreasing this threshold will include reads with a less than optimal match to a library entry which can be helpful to increase sensitivity or control for poor sequence quality.
Ambiguity Threshold (default = 2):
Minimum tolerated difference between primary (best) and secondary (second-best) alignment to consider a read successfully matched. Reads with a difference lower than this threshold will be considered ambiguous and discarded. Increasing this threshold increases stringency. Decreasing this threshold increases sensitivity. With a threshold of 0, the program will accept reads even if they match multiple library entries equally well.
Seed Length (default = 11):
Seed length parameter for Bowtie2 alignment (-L, refer to the Bowtie2 manual for more details). Changing this parameter is generally not required.
Seed Number (default = 1):
Number of allowed mismatches for Bowtie2 seed alignment (-N, refer to the Bowtie2 manual for more details). Changing this parameter is generally not required.
Interval Function (default = "S,1,0.75"):
Bowtie2 seed interval function (-i, refer to the Bowtie2 manual for more details). Changing this parameter is generally not required.
Plotting
Show Non-Targeting Controls (default = Yes):
Highlight non-targeting control sgRNAs (if present in the library) in scatterplots.
Annotate sgRNAs (default = No):
Display sgRNA IDs when highlighting individual genes in scatterplots.
Transparency Level (default = 0.05):
Transparency of points in scatterplots. A lower level is helpful to visualize differences in density.
Dotsize (default = 10):
Size of dots in scatterplots.
PNG Resolution (default = 300):
Resolution for PNG output (dpi).
Spreadsheet Format (default = Excel):
File format for sgRNA and gene tables in the download archive. Use "Excel" to have the workflow automatically convert all text tables into .xlsx format. "Text only" produces only .txt files which saves computation time, but requires manual import into Excel.
AutoHighlight Top Hits (default = Yes): ** NEW in v2.9 ***
Automatically draws scatterplots highlighting the 10 top ranked genes (only available in the download zip file, not on the website). Setting this to "No" saves computation time.
Include Density Plot (default = Yes): ** NEW in v2.9 ***
Draw a density plot of the sgRNA abundance for each sample. Setting this to "No" saves computation time.
Signif. Level (Model Selection) (default = 0.05):
Significance level for choosing a statistical model for the read count distribution. If the data differs significantly from a Poisson distribution, the Negative Binomial distribution is chosen.
1.2.2 Uploading a custom library:
Prepare your library file (e.g. in Excel) as a spreadsheet with 3 columns (with headers):
- gene: This column contains an identifier of the gene that is targeted by the sgRNA
- sgRNA_ID: This column contains an identifier of the sgRNA
- sequence: This column contains the (typically 20bp) sequence of the sgRNA
You can choose other header names for these columns. See example below.
Example:| gene_ID | sgRNA_ID | Seq |
| A1BG | CustomLib00001 | GTCGCTGAGCTCCGATTCGA |
| A1BG | CustomLib00002 | ACCTGTAGTTGCCGGCGTGC |
| A1BG | CustomLib00003 | CGTCAGCGTCACATTGGCCA |
| A1CF | CustomLib00004 | CGCGCACTGGTCCAGCGCAC |
| A1CF | CustomLib00005 | CCAAGCTATATCCTGTGCGC |
| A1CF | CustomLib00006 | AAGTTGCTTGATTGCATTCT |
Save the spreadsheet as either tab-separated format (.tsv) or comma-separated format (.csv). You can use the "Save As" menu item in Excel to do so.
When your file is ready, use the file browser to select and upload your library file.
Next, specify the following parameters:
5’-adapter:
Enter the sequence of the 5’-adapter. Adapters are simply sequences lying 5’ or 3’ of the 20bp sgRNA. There are no restriction to length of your adapter definition, but it is generally recommended to define the 20-25 bp immediately 5’ of the sgRNA sequence (see image below). Also, it is recommended to let the adapter sequence end in an ‘N’ to allow possible mismatches (see example below). A sequence mapping program like SnapGene Viewer is helpful to define the adapter. Definition of the 3’ adapter is not necessary.
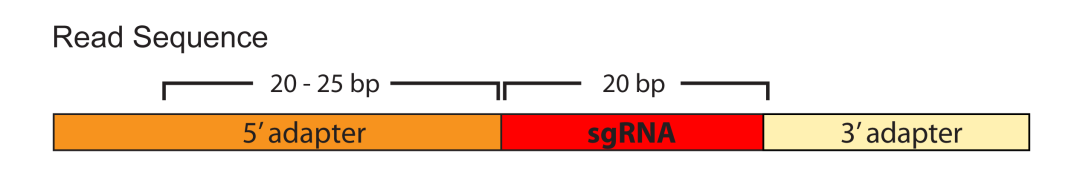
Example: If your reads have the following structure
TCGAATCTTGTGGAAAGGACGAAACACCG ACGGAGGCTAAGCGTCGCAA GTTTTAG
you can, for example, define TCTTGTGGAAAGGACGAAACACCN as the 5’-adapter.
Identifier for non-targeting controls:
If your library contains non-targeting controls, enter an identifier in the library spreadsheet to define the corresponding sgRNAs sequences. The identifier is a part of the gene_ID that is unique to the non-targeting controls (see example below). If your library does not contain non-targeting controls, enter “none”
| gene_ID | sgRNA_ID | Seq |
| Non_Target_0001 | CustomLib34556 | ACGGAGGCTAAGCGTCGCAA |
| Non_Target_0002 | CustomLib34557 | CGCTTCCGCGGCCCGTTCAA |
| Non_Target_0003 | CustomLib34558 | ATCGTTTCCGCTTAACGGCG |
The identifier in this case would be “Non_Target”.
Number of sgRNAs per gene:
Specify the number of sgRNAs targeting a single gene (e.g. 6 in case of the GeCKO library). If this number varies across the library, choose the number that applies to the majority of library entries. Non-targeting controls, miRNAs or other control entries in the library are excluded.
2 Description of the PinAPL-Py Analysis output
The PinAPL-Py output is structured by logical order into tabs and subtabs on the results page. In addition, all output can be downloaded via the “Download Results” button as a single .zip file. Images are saved both as high-resolution .png as well as as .svg vector graphics which can be further processed in Adobe Illustrator or similar image processing software. Tables are saved as raw text (.txt), but can be manually opened with Excel and saved as Excel spreadsheets. For convenience, PinAPL-Py can convert tables on-the-fly (see the “Table Format” parameter on the configuration page), at the cost of additional computation time. NOTE for Windows users: To view text files (.txt/.tsv/.csv), Notepad++ is recommended
NOTE: When the analysis is run with two or more replicate samples for a condition, PinAPL-Py will show an additional sample for that condition (named "<condition name>_avg") where results are averaged across the replicates.
2.1 Gene Ranking Results
Gene Rankings
This tab contains the results of the gene ranking analysis in a sortable table. The columns are:
- Gene: Name of gene (as defined in the library file)
- Gene Score: Value of the computed gene metric score
- Gene Score p-value: Estimated (one-sided) p-value of the gene score, based on a permutation test where sgRNAs are randomly assinged to genes.
- Significant: Statistical significance of the gene score.
- # sgRNAs:Number of sgRNAs targeting the gene
- # Signif. sgRNAs: Number of sgRNAs targeting the particular gene that reached statistical significance in the sgRNA ranking
By default, the table is sorted by p-value. You can sort by other columns (ascending or descending) by clicking on the respective column headers (arrows).
Scatterplots
This tab plots the gene score for each gene (alphabetically sorted on the x-axis). Significant scores are plotted in green. Non-targeting controls (if present) are plotted in orange. The selector at the top can be used to highlight a particular gene of interest (After clicking the selection box, you can type first letter of the gene name to find the gene more quickly).
p-Value Distribution
This tab shows the distribution of p-values from the gene ranking analysis. Typically, a bimodal distribution is seen, with a small bar on the left end and a large bar on the right end.
sgRNA Efficacy:
This tab shows information about the overall efficacy of sgRNAs targeting the same gene. Genes are categorized by the number of targeting sgRNAs that reached statistically significant enrichment/depletion. Genes having no significant sgRNAs are omitted.
2.2 sgRNA Ranking Results
Rankings
This tab contains the results of the sgRNA ranking analysis in a sortable table. The columns are:
- sgRNA: Identifier of sgRNA
- Gene: Name of target gene
- Counts: Normalized read count
- Control Mean: Average normalized read count of this sgRNA in the control
- Control StDev: Standard deviation of normalized read count in the control
- Fold Change: The ratio of normalized read count in the sample to the control average
- p-value: p-value (one-sided) of the enrichment/depletion of this sgRNA compared to the control
- Significant: Statistical significance of the enrichment/depletion of this sgRNA
By default, the table is sorted by p-value. You can sort by other columns (ascending or descending) by clicking on the respective column headers (arrows).
Plots
Treatment vs Control
Scatterplots of normalized sgRNA read counts in the sample versus the average normalized count in the controls. The fraction reaching significant enrichment/depletion (dependent on screen type) compared to the control is plotted in green. Non-targeting controls (if present) are plotted in orange. The selector at the top can be used to highlight a particular gene of interest (After clicking the selection box, you can type first letter of the gene name to find the gene more quickly). After highlighting a particular gene, the IDs of the corresponding sgRNAs can be displayed using the checkbox on the far right. Highlighting of non-targeting controls can be switched on or off with the checkbox next to the gene selector.
Density Plots **** NEW in v2.9 ***
Same data as the previous tab, but showing a density plot of the sgRNA abundance.
Volcano Plots
This tab plots sgRNA p-value against fold-change. The fraction reaching significant enrichment/depletion (dependent on screen type) compared to the control is plotted in green. Non-targeting controls (if present) are plotted in orange. The selector at the top can be used to highlight a particular gene of interest (After clicking the selection box, you can type first letter of the gene name to find the gene more quickly). After highlighting a particular gene, the IDs of the corresponding sgRNAs can be displayed using the checkbox on the far right. Highlighting of non-targeting controls can be switched on or off with the checkbox next to the gene selector. p-values are capped at 1e-16 for technical purposes.
z-Score Plots
This tab shows the fold-change for each sgRNA ranked from lowest to highest. The z-Score is the normalized deviation from the mean read count. The fraction reaching significant enrichment/depletion (dependent on screen type) compared to the control is plotted in green. Non-targeting controls (if present) are plotted in orange. The selector at the top can be used to highlight a particular gene of interest (After clicking the selection box, you can type first letter of the gene name to find the gene more quickly). After highlighting a particular gene, the IDs of the corresponding sgRNAs can be displayed using the checkbox on the far right. Highlighting of non-targeting controls can be switched on or off with the checkbox next to the gene selector.
p-Values
This tab shows the distribution of p-values from the sgRNA enrichment/depletion analysis.
Read Count Distribution
This tab shows information about the statistical distribution of sgRNA read counts.
- Left: Lorenz curves and Gini coefficients: The Lorenz curve visualizes the distribution of reads, showing what fraction of sgRNAs (green) or genes (blue) is represented by what fraction of reads. The Gini coefficient quantifies the difference of this distribution from a perfectly even distribution. A perfectly even distribution would result in a diagonal curve (Gini coefficient = 0) and would indicate a complete absence of selection in the screen. An extremely uneven distribution results in a flat curve (Gini coefficient = 1) and would indicate extreme selection (only a single sgRNA/gene is represented in the sequencing data). Thus, the more selective the conditions of the screen are, the closer will the Gini coefficient approach 1.
- Right: Boxplots and histograms: Boxplots and histograms for the read counts per sgRNA (green) or gene (blue), respectively. Outliers are omitted for visualization purposes.
- Bottom: Summary: Descriptive statistics are summarized. sgRNA/gene representation measures the number of sgRNAs/genes detected by at least one read count in the sample (as percentage of the full library).
Clusters
This tab shows a cluster analysis (heatmap) of all samples in the dataset, based on the most variable or most abundant/depleted sgRNAs (as chosen on the configuration page). Log10 normalized read counts are color-coded from lowest (yellow) to highest (red).
Replicate Correlation:
Scatterplots showing the normalized sgRNA read counts in one replicate of each condition versus another. Pearson and Spearman correlation coefficients are reported. Non-targeting controls (if present) are plotted in orange.
Read Count Dispersion:
This tab shows the distribution of read counts in the control samples only. These data are used to estimate the parameters of the statistical model describing the distribution of sgRNA read counts throughout the rest of the dataset.
- Left: Read Count Overdispersion: This plot visualizes the degree of overdispersion in the data, i.e. the degree by which the variance of read counts exceeds the mean (as typically seen in next-generation sequencing datasets). In case of significant overdispersion, a negative binomial model is chosen over a Poisson model.
- Right: Mean/Variance Model: This plot shows shows a regression of log overdispersion against log mean. This is required to compute the parameters of the statistical model.
2.3 Alignment Results
Summary
This tab shows the sequencing depth (number of total reads) per sample as well as the fractions of reads successfully or unsuccessfully aligned to the reference library. Alignment Unique: Only a single match found in the reference library. Alignment Tolerated: Multiple matches found in the reference library, but the difference in matching score between the best and second-best was above the ambiguity threshold. Alignment Ambiguous: Multiple matches found in the reference library, and the difference in matching score between the best and second-best was below the ambiguity threshold. Alignment Failed: No match found in reference library. Reads with failed and ambiguous alignment are discarded prior to the subsequent enrichment/depletion analysis.
Alignment Statistics
- Left: Mapping Quality: Histogram of the overall quality by which the reads mapped to the reference library. Reads that uniquely align to a single library sequence yield a high mapping quality score. Reads that ambiguously align to multiple library sequences or that do not align to any library sequence yield a low mapping quality score. For more detailed information about computation of the mapping quality score, please refer to the Bowtie2 manual.
- Right: Alignment Analysis: Barplot showing the primary (best) and secondary (second-best) alignment scores achieved for each read. If a read perfectly aligns to only one library sequence, its primary alignment score will be maximal, and its secondary alignment score will be 0. If a read aligns ambiguously to multiple library sequences, its secondary alignment score will be close to its primary alignment score. If a read does not align to any library sequence, both its primary and secondary alignment scores will be 0. The fraction of reads marked in red is being discarded prior to enrichment/depletion analysis.
- Bottom: Summary: This text summarizes the the success of the alignment. For an explanation of the parameter settings reported at the bottom of the page, see the ALIGNMENT section).
Sequence Quality:
This tab provides analyses for sequence quality control (produced by fastqc). For the full fastqc output, click the “See full report” link
- Upper Left: Per Base Quality: This plot shows the quality distribution for every base position in the read. y-axis is sequence quality score (Phred). Preferably, the majority of the read is in the green area.
- Upper Right: Per Sequence Quality: This plot shows a sequence quality histogram. y-axis shows number of reads. Preferably, sequence quality should peak at a score >= 35.
- Lower Left: GC Content: This plot shows a histogram of the the GC content. y-axis shows number of reads.
- Lower Right: Per Base Sequence Variation: This plot shows the fractions of T, C, A and G for every base position in the read. A balanced mix is typically only seen in the 20 bp sgRNA sequence.
2.4 Run Info
Output Log
This shows the program execution log. If you experience technical difficulties during your run, browse the log for error messages as they can provides clues for trouble-shooting.
Configuration
This file shows the parameter settings used for the analysis run.
Sample Names
This table shows file names and the corresponding sample names. Replicates of the same condition are numbered automatically.
3 REFERENCES
Anders,S. and Huber,W. (2010) Differential expression analysis for sequence count data. Genome Biol. , 11 , R106.
Doench,J.G. et al. (2016) Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. , 34 , 184 –191.
Li,W. et al. (2014) MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. , 1 –12.